
Clinical trial blinding is a procedure in which one or more parties in a trial are kept unaware of which treatment arms participants have been assigned to. Per ICH E9 (4) guidelines, blinding is intended to limit conscious and unconscious bias in the design, conduct, analysis, and interpretation of clinical trial results.
Clinical trial sponsors follow controlled processes to prevent information directly, or indirectly informing a party of the treatment given to a subject. Moreover, the use of advanced technology – interactive response technology (IRT) – further reduces the potential for bias, specifically during randomization and trial supply management (RTSM). However, there are still areas where risks of unintentional unblinding arise – and minimizing those risks is of paramount importance.
How Unintentional Unblinding Occurs in Clinical Trials
There are numerous trigger points that may result in unintentional unblinding during clinical trial execution including when the following scenarios occur:
Randomization
- Reporting randomization number when blocks are dynamically allocated to strata (with a higher risk with site stratification)
- Reporting randomization number when forced randomization is permitted (with a higher risk with site stratification and if there are only two treatment arms)
- What investigational medical product (IMP) must be available at the site before randomization is permitted to proceed, usually referred to as medication checks done by an IRT (the risk depends on the number of treatment arms and also whether forcing is permitted)
- Reporting randomization number when patients are being replaced
- If re-randomization is required for a sub-set of treatment arms
Dispensing
- What IMP must be available at the site before medication assignment is permitted to proceed (the risk depends on the number of treatment arms)
- The mechanism for selecting which packs should be dispensed to patients from site stocks
- When dispensing only a partial amount of required treatment (which could be allowed if the patient can be given sufficient medication until the remainder can be provided)
- When blinded medication is used in an open-label manner (i.e., single-blind run-in)
- When the dose level is known to investigators, but the treatment is not
- When one or more treatments are supplied by the site (i.e., the IRT only manages the ‘active’ pack, with the comparator supplied by the site)
- If third-party lab data is required to make a patient treatment decision (see figure 1)
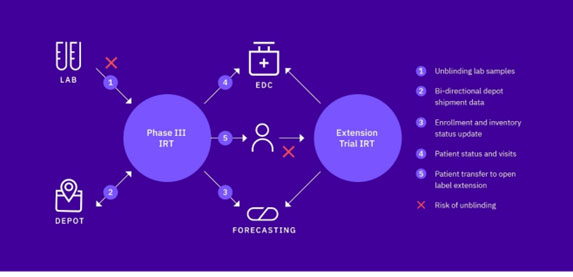
Shipping
- Selecting which packs should be included in shipments
- If a partial shipment is sent to a site due to a stock-out at a depot
- When shipping in boxes, but dispensing individual packs to patients (the risk depends on the design of the pack number and whether the box will be visible to blinded staff during or after dispensation at the site)
- When different expiry dates are used across separate batches of different blinded pack types
- If there are different trigger and resupply values (buffer stock) for different IMP
- If the initial shipment to the site only contains one of each IMP and like-for-like replacement shipments are allowed following each single pack allocation
Clinical Trial Unblinding: What it is and what it isn’t
It’s well accepted that unintentional unblinding can have ramifications for the study data as well as for individual patients. But, while there are absolutes on what unblinding is – such as revealing the treatment assignment on a confirmation intended for blinded staff – there are certain aspects that are a little less clear and open for debate and interpretation, including the following edge issues:
- Is there a difference between knowing the treatment assignment of an individual subject vs. a group of subjects in an open-label study? Does aggregate data lead to the same bias in an open-label study as it would in a blinded study?
- What about random allocation of supplies vs a predetermined dispensing sequence? Can we protect the blind better through this process?
- Are randomly generated container / pack numbers an added safety net for unblinding or are they reducing ease of use for site staff?
- What are the perspectives on not assigning a pack (post-randomization) if not all material types are available?
Partial Unblinding
Questions also exist related to partial unblinding. Is it a real concern or an overreaction? Is there data to suggest it has a real-world impact or is it just a perception?
Partial unblinding can become full unblinding if one patient code is broken, for example when it is known that:
- Two patients are on the same treatment arm, or two packs contain the same medication
- Two patients are on different treatment arms, or two packs contain different treatments
As IRT technology and our approaches to RTSM advance, it’s becoming more important than ever that we continue to focus our efforts on blinding considerations and taking the necessary steps to ensure the risk of unintentional unblinding is mitigated at every step of the clinical development process. Working with a reliable provider of robust IRT solutions can go a long way toward reducing the risk of unintentional unblinding and maintaining the integrity of regulated clinical trials.







